
概要・定義
高フェニルアラニン血症(HPA)は、L-フェニルアラニン(Phe)をL-チロシン(Tyr)に変換するフェニルアラニン水酸化反応の異常により血漿中Phe値が2mg/dlを越える状態で、その原因によりフェニルアラニン水酸化酵素(PAH)の異常とその補酵素のテトラヒドロビオプテリン(BH4)の代謝異常に分類される。(BH4欠損症は別項参照)。PKUは、診断時の血漿中Phe値により古典的PKU(20mg/dl以上)、軽症PKU(10mg/dl以上20mg/dl 未満)、軽症HPA(10mg/dl未満)に分類されている。BH4反応性PAH欠損症は、PKUと同じPAHの異常であるが補酵素BH4に反応して血中Phe値が低下するためBH4反応性PKU やBH4反応性HPAとよばれている。いずれも発症前に治療を開始すれば予後は良好である。
疫学
頻度は古典的PKUが約9万人に1例、HPAが約16万人に1例で、両者をあわせるとPAHの異常によるHPAの頻度は約7.6万人に1例となる。BH4反応性PAH欠損症の頻度はPAH欠損症の約25〜40%程度と推測される。
病因
フェニルケトン尿症(PKU)は12番染色体(12q23.2 )に位置するPAH遺伝子(PAH)の変異に基づく遺伝性疾患で常染色体劣性遺伝形式をとる先天代謝異常症である。
症状
古典的PKUでは無治療の場合、高Phe血症による精神発達遅延やチロシンの低下による色白や赤毛などの色素欠乏の症状を呈する。
診断
新生児マススクリーニングで高Phe血症で発見された場合、血液・尿プテリジン分析と乾燥濾紙血ジヒドロプテリジン還元酵素(DHPR)活性の測定により補酵素BH4の鑑別診断を行う。
治療
古典的PKUはPhe制限食により、BH4反応性PKUはBH4の投与により血中Phe値を下記の維持範囲にコントロールする。
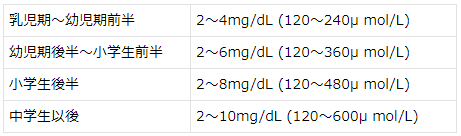
血中Phe値の維持範囲1
予後
乳児期早期より治療を開始し血中Phe値をコントロールすれば全く正常に発達し、予後は良好である。
成人期以降
女性患者が妊娠する場合は妊娠前から血中Phe値を5mg/dL程度にコントロールし出産まで維持することで母性PKUを予防することができる2。
参考文献
1) 北川照男、松田一郎、青木菊麿、他:フェニルケトン尿症(高フェニルアラニン血症の一部を含む)治療指針の 第 2 次改定の経緯と改定勧告治療指針(平成 24 年度)について、特殊ミルク情報第2012;48号、82-84
2) 特殊ミルク共同安全開発委員会: アミノ酸代謝異常症のために、食事療法ガイドブック。 恩賜財 団母子愛育会 2008
- 版
- :バージョン2.0
- 更新日
- :2015年5月25日
- 文責
- :日本先天代謝異常学会
