
概念・定義
Alport症候群(AS)は1927年にAlportにより報告された進行性の腎炎を特徴とする遺伝性疾患で,聴力異常・眼球異常の合併を特徴とする(1)。その原因遺伝子は,COL4A3,COL4A4,COL4A5遺伝子である。
1990年に初めてCOL4A5遺伝子変異がX染色体連鎖型(X-linked Alport syndrome: XLAS)家系で同定され(2),次いで1994年にCOL4A3あるいはCOL4A4遺伝子変異が常染色体劣性型(autosomal recessive Alport syndrome: ARAS)家系で同定されている(3)。AS全体の中で,XLASが約80%,ARASが約15%を占める.残りの5%の中にCOL4A3あるいはCOL4A4遺伝子の異常から引き起こされる常染色体優性型が報告されている(4)。
ASによる糸球体腎炎は糸球体基底膜(GBM)の菲薄化・断裂を伴って進行し,数年から十数年を経て慢性腎不全に至る(5)。
初期症状は無症候性血尿であることが多く,特にXLASの男性において成人になってからほぼ例外なく腎不全に至る。
XLASのキャリアである女性のASによる糸球体腎炎は,無症候性血尿のみの症例から早期に慢性腎不全に至る症例まで多様であり,X染色体の不活化現象によると推測されている(5)。
ASによる糸球体腎炎は糸球体基底膜(GBM)の菲薄化・断裂を伴って進行し,数年から十数年を経て慢性腎不全に至る(5)。
初期症状は無症候性血尿であることが多く,特にXLASの男性において成人になってからほぼ例外なく腎不全に至る。
XLASのキャリアである女性のASによる糸球体腎炎は,無症候性血尿のみの症例から早期に慢性腎不全に至る症例まで多様であり,X染色体の不活化現象によると推測されている(5)。
病因・病態
ASの原因遺伝子であるCOL4A3,COL4A4,COL4A5のいずれの遺伝子に変異がある場合にも,IV型コラーゲンα3鎖,α4鎖およびα5鎖による3重らせん構造が適切に形成されないため,腎炎を引き起こすと考えられている。
腎炎に関しては,GBMの構造が脆弱となり,腎障害を引き起こすと考えられている。
腎炎に関しては,GBMの構造が脆弱となり,腎障害を引き起こすと考えられている。
診断・鑑別診断
ASの有病率は欧米で約1:5,000と報告されている。本邦では学校検診などで無症候性血尿を契機に発見されることが多い。そのため,無症候性血尿の症例に対して,ASを疑って腎疾患の家族歴を聴取することが重要である(表1)(6)。
厚生労働科学研究費補助金(難治性疾患等政策研究事業)「腎・泌尿器系の希少・難治性疾患群に関する診断基準・診療ガイドラインの確立」班により平成27年に改訂されたASの診断基準を表2に示す(7)。
家族歴からASが疑われる場合は腎生検を行うことが望ましい。一方,10歳以下の症例では血尿が唯一の症状であることが多く,腎不全の家族歴が明らかでない場合鑑別は困難である。家族性に血尿がみられるが腎不全の家族歴がない場合,その血尿患者の腎生検の適応は通常の腎生検の適応と同じである(8, 9)。
電子顕微鏡で特徴的な糸球体基底膜の広範で不規則な肥厚と徹密層の網目状変化を認め,これにより確定診断できる。非薄化・断裂が認められる場合もある。ただし,良性家族性血尿においてしばしばみられる糸球体基底膜の広範な非薄化も本症候群においてみられることがあり,注意を要する。
糸球体基底膜の厚さは正常では300 nm以上であるが,良性家族性血尿や本症候群では150~200nmと異常に薄い場合がみられ,糸球体基底膜が断裂して糸球体上皮細胞と内皮細胞が直接接触していることもある。本症候群の糸球体基底膜は,網目状変化のために肥厚した部分,異常に薄い部分,正常な部分が混在する。
確定診断の一助として,腎組織でのIV型コラーゲンα3,4,5鎖タンパクに対するモノクローナル抗体を用いた免疫染色も有効である(10)。
典型的には,XLASの男性ではGBMでIV型コラーゲンα3,4,5鎖タンパクの発現は消失し,XLASの女性ではGBMでこれら3つのタンパクはすべてモザイク状となる。また,ARASの男性・女性ともIV型コラーゲンα3,4,5鎖タンパクは消失する。ただし,これらのタンパク発現が正常でも本症候群は否定できない。
最も確実なASの診断は,遺伝子解析である。α3~α5鎖遺伝子の変異が検索され,現在は解析法の進歩によりほぼ100%原因遺伝子変異が検出できる。ただし,遺伝子検査が可能な施設が限られることや,コストパフォーマンスを考慮した場合に診断法の第一選択にはならない。
表1 Alport症候群の診断(6)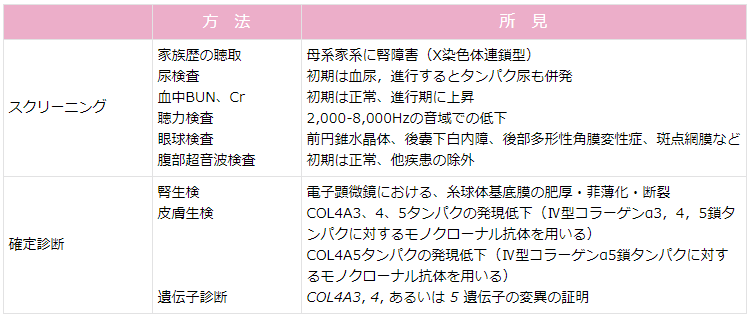 表2 アルポート症候群診断基準 (平成27 年2 月改訂)
厚生労働科学研究費補助金(難治性疾患等政策研究事業)「腎・泌尿器系の希少・難治性疾患群に関する診断基準・診療ガイドラインの確立」班
表2 アルポート症候群診断基準 (平成27 年2 月改訂)
厚生労働科学研究費補助金(難治性疾患等政策研究事業)「腎・泌尿器系の希少・難治性疾患群に関する診断基準・診療ガイドラインの確立」班

厚生労働科学研究費補助金(難治性疾患等政策研究事業)「腎・泌尿器系の希少・難治性疾患群に関する診断基準・診療ガイドラインの確立」班により平成27年に改訂されたASの診断基準を表2に示す(7)。
家族歴からASが疑われる場合は腎生検を行うことが望ましい。一方,10歳以下の症例では血尿が唯一の症状であることが多く,腎不全の家族歴が明らかでない場合鑑別は困難である。家族性に血尿がみられるが腎不全の家族歴がない場合,その血尿患者の腎生検の適応は通常の腎生検の適応と同じである(8, 9)。
電子顕微鏡で特徴的な糸球体基底膜の広範で不規則な肥厚と徹密層の網目状変化を認め,これにより確定診断できる。非薄化・断裂が認められる場合もある。ただし,良性家族性血尿においてしばしばみられる糸球体基底膜の広範な非薄化も本症候群においてみられることがあり,注意を要する。
糸球体基底膜の厚さは正常では300 nm以上であるが,良性家族性血尿や本症候群では150~200nmと異常に薄い場合がみられ,糸球体基底膜が断裂して糸球体上皮細胞と内皮細胞が直接接触していることもある。本症候群の糸球体基底膜は,網目状変化のために肥厚した部分,異常に薄い部分,正常な部分が混在する。
確定診断の一助として,腎組織でのIV型コラーゲンα3,4,5鎖タンパクに対するモノクローナル抗体を用いた免疫染色も有効である(10)。
典型的には,XLASの男性ではGBMでIV型コラーゲンα3,4,5鎖タンパクの発現は消失し,XLASの女性ではGBMでこれら3つのタンパクはすべてモザイク状となる。また,ARASの男性・女性ともIV型コラーゲンα3,4,5鎖タンパクは消失する。ただし,これらのタンパク発現が正常でも本症候群は否定できない。
最も確実なASの診断は,遺伝子解析である。α3~α5鎖遺伝子の変異が検索され,現在は解析法の進歩によりほぼ100%原因遺伝子変異が検出できる。ただし,遺伝子検査が可能な施設が限られることや,コストパフォーマンスを考慮した場合に診断法の第一選択にはならない。
表1 Alport症候群の診断(6)
表2 アルポート症候群診断基準 (平成27 年2 月改訂)
厚生労働科学研究費補助金(難治性疾患等政策研究事業)「腎・泌尿器系の希少・難治性疾患群に関する診断基準・診療ガイドラインの確立」班
治療・予後
ASに対する根治的治療法は現在のところ存在せず腎保護療法が行われる。アンジオテンシン変換酵素(ACE)阻害薬やアンジオテンシン受容体拮抗薬(ARB)を第一選択薬とする(11)。
腎障害が進行し,慢性腎不全に至った場合の治療法としては,透析療法(血液透析・腹膜透析)と腎移植に分けられる。移植後抗基底膜抗体腎炎を発症する可能性があるものの,腎移植の正着率は比較的良好であると報告されており(11),若年で慢性腎不全に陥った症例に対してはその適応が考慮される。
腎障害が進行し,慢性腎不全に至った場合の治療法としては,透析療法(血液透析・腹膜透析)と腎移植に分けられる。移植後抗基底膜抗体腎炎を発症する可能性があるものの,腎移植の正着率は比較的良好であると報告されており(11),若年で慢性腎不全に陥った症例に対してはその適応が考慮される。
参考文献
1) Alport AC: Hereditary familial congenital haemorrhagic nephritis. Br Med J 1:504-506, 1927
2) Barker DF, Hostikka SL, Zhou J, et al: Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 248:1224-1227,1990.
3) Mochizuki T, Lemmink HH, Mariyama M, et al. Identification of mutations in the alpha 3(IV) and alpha 4(IV) collagen genes in autosomal recessive Alport syndrome. Nat Genet 8:77-81, 1994
4) Jefferson JA, Lemmink HH, Hughes AE, et al:Autosomal dominant Alport syndrome linked to the type lV collage alpha 3 and alpha 4genes(COL4A3 and COL4A4).Nephrol Dial Transplant 12:1595-1599, 1997
5) Jais JP, Knebelmann B, Giatras I, et al:X-linked Alport syndrome:natural history and genotype-phenotype correlations in girls and women belonging to l95 families:a "European Community Alport Syndrome Concerned Action',study. J Am Soc Nephol 14:2603-2610, 2003
6) 片山 鑑,堅村信介:Alport症候群,p233-235,南江堂,2011.
7) 厚生労働科学研究費補助金(難治性疾患等政策研究事業)「腎・泌尿器系の希少・難治性疾患群に関する診断基準・診療ガイドラインの確立」班報告書,日腎誌(印刷中)
8) 日本腎臓学会 編.CKD診療ガイドライン2013. 東京医学社, 東京, 2013
9) 日本腎臓学会 編.CKD診療ガイドライン2009. 東京医学社, 東京, 2009
10) Kashtan CE, Ding J, Gregory M,et al; Alport Syndrome Research Collaborative.Clinical practice recommendations for the treatment of Alport syndrome: a statement of the Alport Syndrome Research Collaborative. Pediatr Nephrol 28:5-11, 2013
11) Kashtan CE. Alport syndrome. An inherited disorder of renal, ocular, and cochlear basement membranes. Medicine 78:338-360, 1999
- 版
- :バージョン1.1
- 更新日
- :2015年3月30日
- 文責
- :日本小児腎臓病学会
